At UGA you can run AlphaFold on two platforms, GACRC and COLAB. The code on GACRC might be closer to the original one released by Deepmind. The COLAB one made the ‘sequence alignment’ step quicker with potentially reduced accuracy. The good news is that the results from both of them look the same to my eyes. After getting the results, just use software like PyMol to open the .pdb file to visualize the 3D model.
EMBL already have a collection of prediction results for a bunch of model organisms here: https://alphafold.ebi.ac.uk/download
AlphaFold2 is just one click away: COLAB
The AlphaFold2 on COLAB is very easy to use through a browser with this link: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb
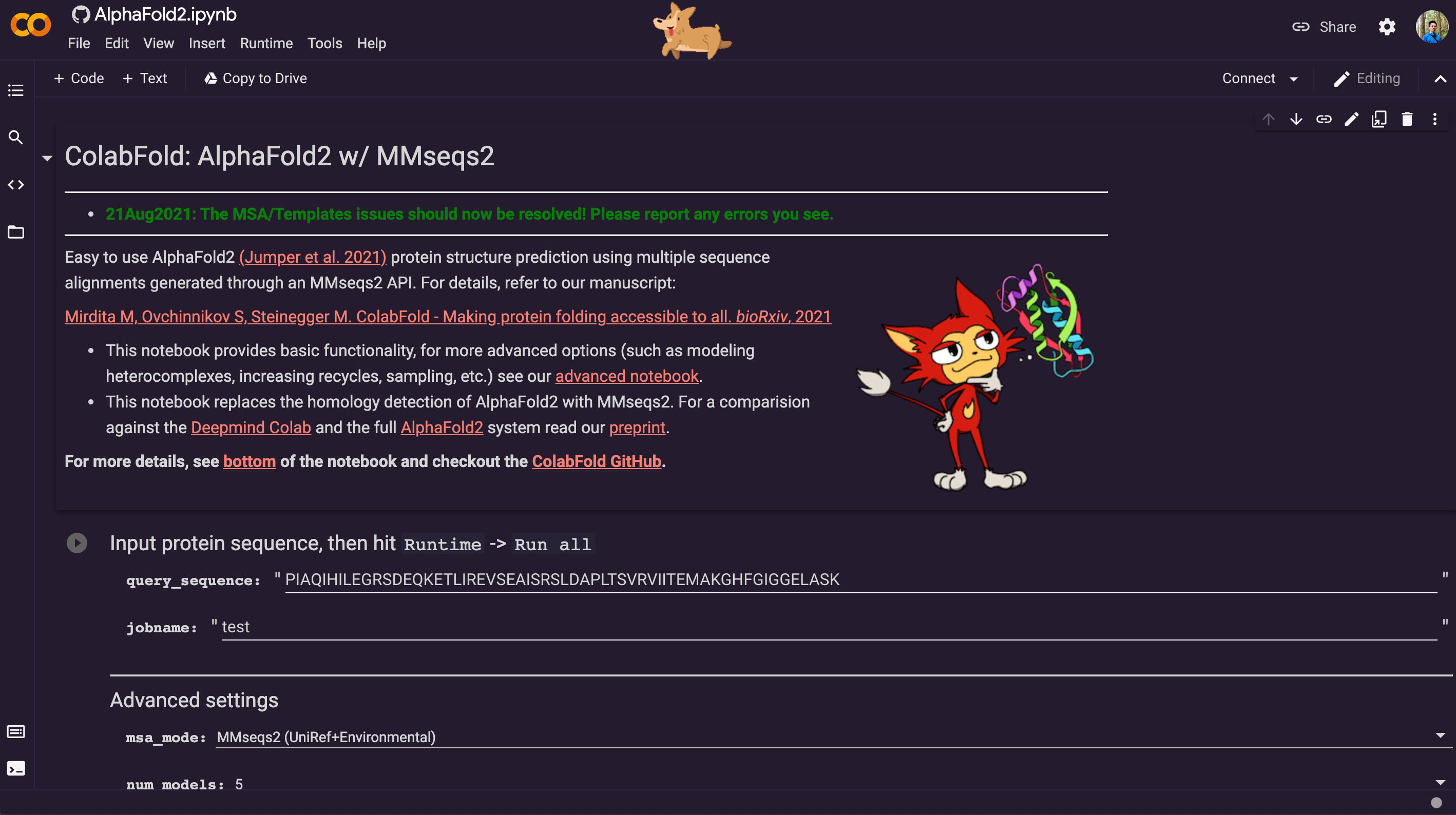
It already has a very decent graphic user interface. Besides potential accuracy loss, the only drawback is you can only submit one sequence at a time through the graphical user interface.
Batch AlphaFold submission to GACRC (WIP)
If you need to submit a bunch of sequences we can do that on GACRC. Document from GACRC about Alphafold2: https://wiki.gacrc.uga.edu/wiki/AlphaFold-Sapelo2
First, Alphafold2 can only run one sequence at a time. So we need to split the batch sequence fasta file into several fasta files that contain only one sequence. Sample code:
from Bio import SeqIO
# seperate sequences into files
file_name_list = []
for seqRecord in SeqIO.parse("Ptrichocarpav4.1g.primaryTrs.pep.fa", "fasta"):
file_name_list.append(f"{seqRecord.id}.fa")
SeqIO.write(seqRecord, f"{seqRecord.id}.fa", "fasta")
# prepare commands in a file for parallel
# command template
# bash $EBROOTALPHAFOLD/alphafold/run_alphafold.sh -d /db/AlphaFold -o ./output/ \
# -m model_1 -f ./{file_name} -t 2020-05-14
with open('alphaFold.cmd', 'w') as f:
for file_name in file_name_list:
cmd = f"bash $EBROOTALPHAFOLD/alphafold/run_alphafold.sh -d /db/AlphaFold -o ./output/ \
-m model_1 -f ./{file_name} -t 2020-05-14"
f.write(cmd)
Then use the following script to submit the job that runs prediction for each sequence in the folder parallelly:
#!/bin/bash
#SBATCH --job-name=alphafold
#SBATCH --partition=gpu_30d_p
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=32
#SBATCH --gres=gpu:P100:1 # must use P100 for the memory space
#SBATCH --mem=180gb
#SBATCH --time=720:00:00 #30*24
#SBATCH --output=%x.%j.out
#SBATCH --error=%x.%j.err
#SBATCH --mail-user=yourEmail@uga.edu
#SBATCH --mail-type=ALL
cd $SLURM_SUBMIT_DIR
ml CUDA
ml AlphaFold/2.0.0_conda
ml parallel
# single execution
# bash $EBROOTALPHAFOLD/alphafold/run_alphafold.sh -d /db/AlphaFold -o ./test/ \
# -m model_1 -f ./3fasta.fa -t 2020-05-14
# parallel execution
parallel --results outdir -j 0 -a alphaFold.cmd