
原文發表於第 11 屆 iT 邦幫忙鐵人賽 (https://ithelp.ithome.com.tw/articles/10221594)
燕尾服套裝軟體取名的惡趣味總讓我想到黑暗大法師
RNA-Seq 主要目的之一就是推估樣本中轉錄產物豐富度,這個過程可以分成
短讀序在參考序列上位置之指定 (read assignment)
依據位置指定結果定量 (quantification)
兩個階段,依據定量結果進行統計檢定,就可以定義出試驗設計造成的差異表現基因,後續銜接其他分析。
上述兩階段的軟體很多,有些只涵蓋其中一步驟,有些單一軟體就處理好兩個步驟。除了使用場景之不同以外,依據軟體實際演算法之不同可以分成基於排比結果 (alignment-based) 與無需排比 (alignment-free) 兩大類。今天先鎖定 alignment-based 工具,明天才會介紹神秘的 alignment-free 工具。Alignment-based 工具可以說是透過回貼短序列來指定位置的列,這個過程也是非常耗時的步驟,一代代的工具推陳出新,每一代都比之前的更快更準,並且已經達到微陣列之精準程度。近幾年的各項軟地位可以說是逐漸穩定,序列比對軟體戰國時代終結。
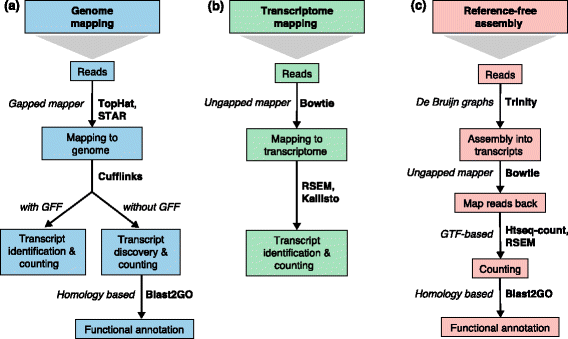
Bowtie (領結)、TopHat (高頂禮帽)、以及 Cufflink (袖扣) 等等幾個以燕尾服配件為名的軟體被合稱做燕尾服套裝 (Tuxedo Suite),後來還有 HISAT2、StringTie (蝶形領結)、Ballgown (舞會禮服) 組合成新一代的燕尾服分析流程。
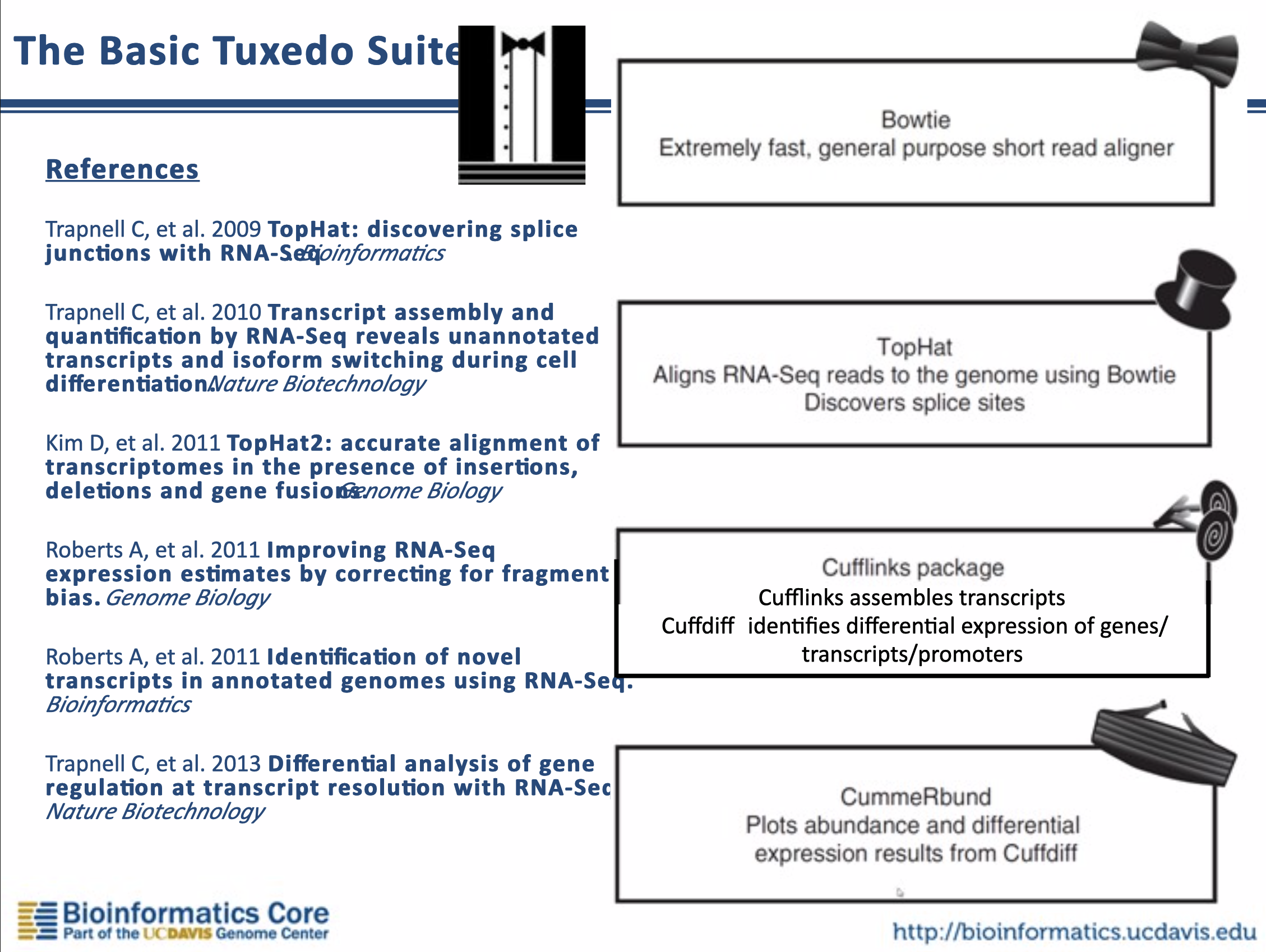
目前的理解是新燕尾服套裝 (HISAT2, StringTie, Ballgown) 就是就燕尾服套裝 (TopHat, Cufflink) 的繼任者,但是 Bowtie2 偶爾還是會在其他適合的場景被使用,因此本文現階段選擇 Bowtie2 簡介 alignment-based mapper 的大概樣貌。
Bowtie2 簡介
選擇 Bowtie2 介紹的另一個原因其實是因為幾年前曾經報告過這個工具的文獻,可以直接用當年的投影片來充版面(X)。序列比對演算法真的是生物資訊中非常令人感興趣的部分,也是眾多工具的根基,但也是最深奧難懂的部分,未來希望可以持續擴充相關的演算法概念介紹。以下內容可能有誤,請不吝指教!
Bowtie2 的功能,就是在蒼蒼的 reference.fasta 中,找到短短的 reads.fastq 之歸屬。逐字比對太慢,所以必須要借用 index 與 dyanmic programming 之力。這也是為什麼大部分的比對軟體都會分成建立 index 與實際比對兩個指令。
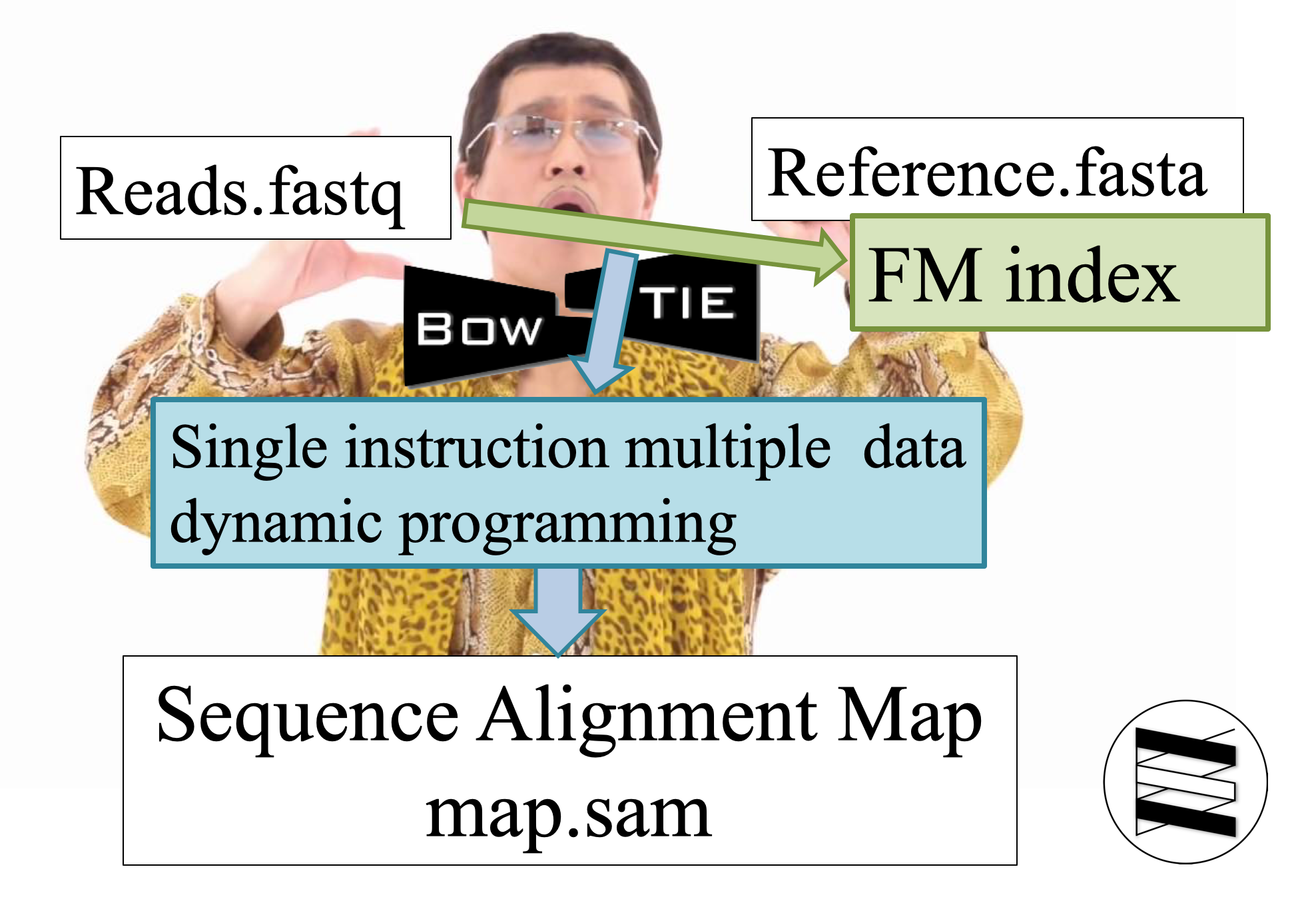
第一步是替 reference.fasta,也就是參考之基因體或轉錄體,建立 FM-index。所謂 FM-index 就是透過 Burrows-Wheeler transform 這個轉換方式,壓縮原本巨大的 reference.fasta。
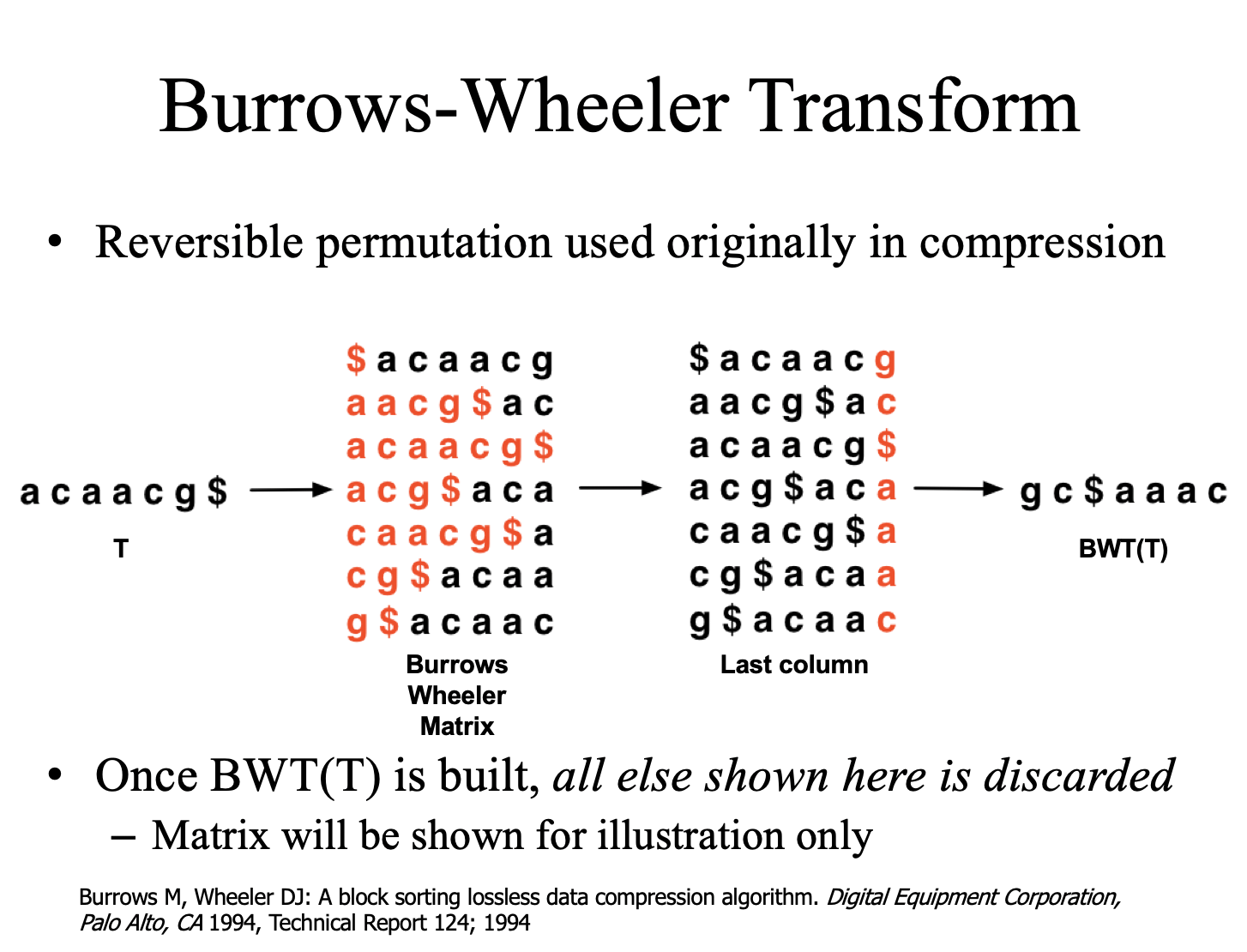
將要壓縮的字串的每個文字輪流排到第一位,其餘照原本的順序排列。長度 n 的字串就有 n 種排列方式,再將第一位的字符依據字母順序縱向排序,形成一個 n*n 的矩陣,之後取最右邊的 column,這就是 BWT 轉換。長度 n 的字串經過 BWT 轉換還是長度 n 的字串,但是經過這樣的轉換之後,相同的字符會比較容易排列在相鄰的位置,連續出現的字符就可以簡寫,達到壓縮的目的。
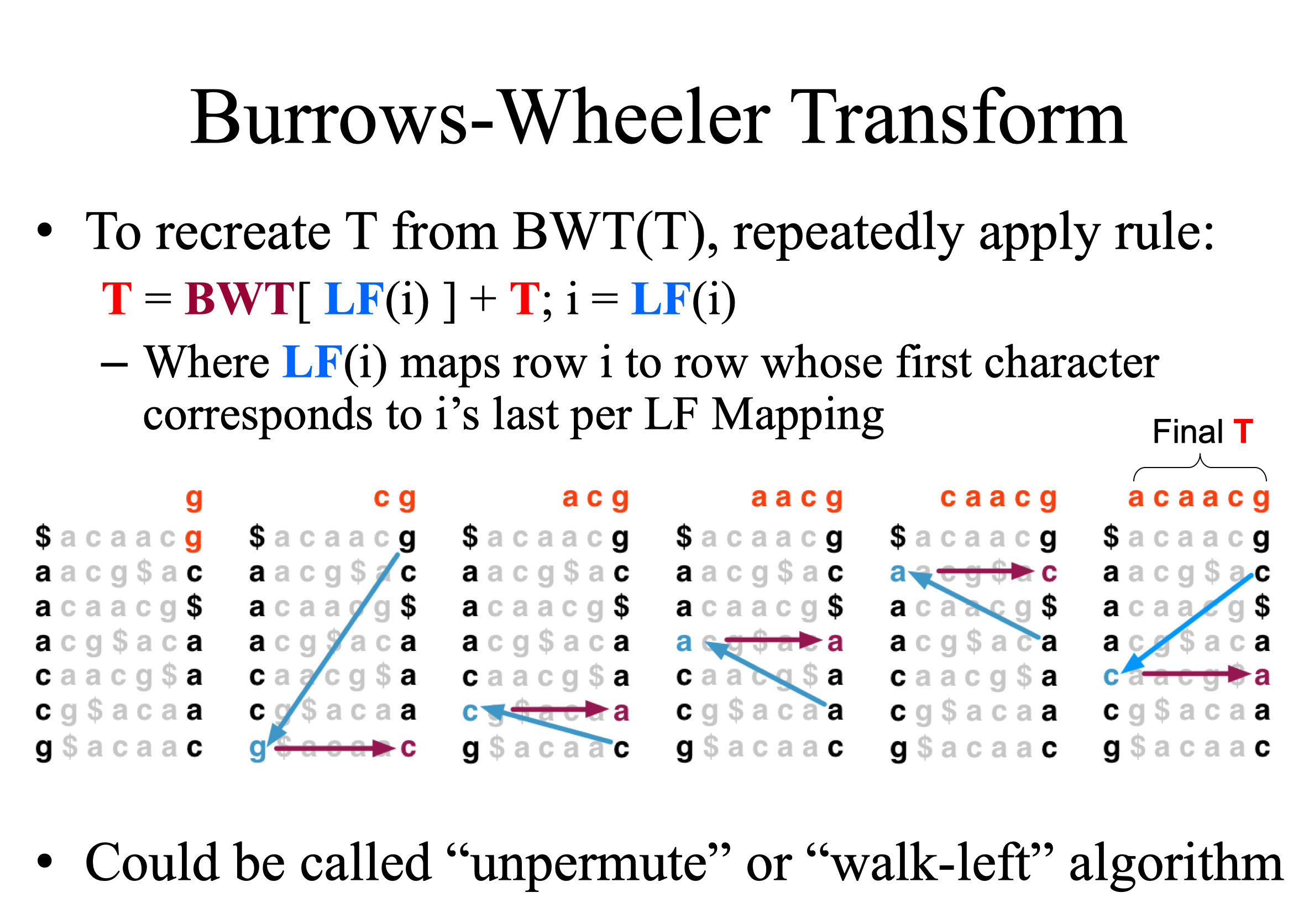
經過 BWT 轉換的字串完全可以逆向回推出原本字串,這個過程稱作 walk-left。
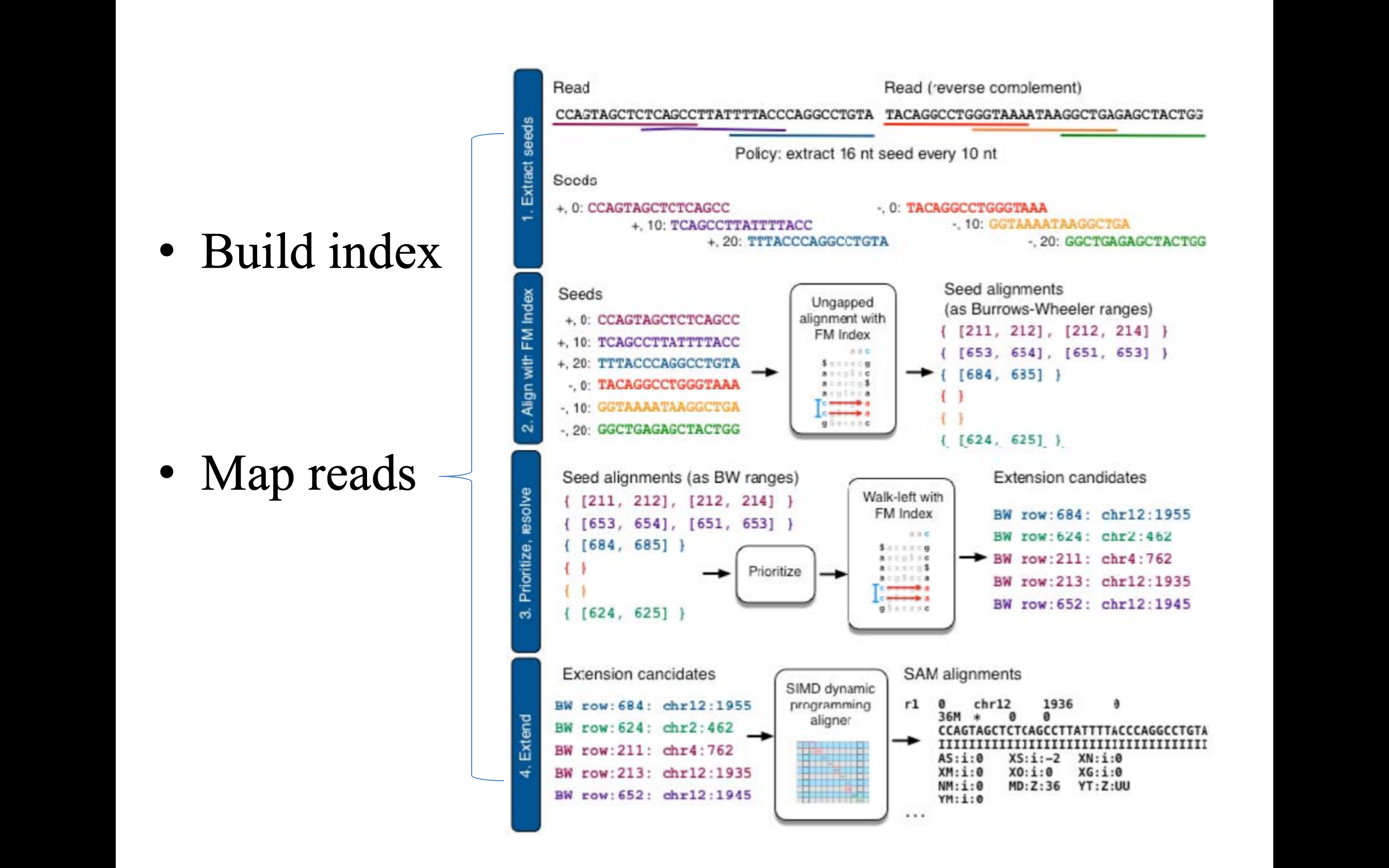
上述的 BWT 轉換建立 FM-index 只是第一步,第二部的 Map reads 則又可以細分成四個步驟:Extract seeds, Align with FM index, Prioritize and resolve, Extend。Extract seeds 只是將要比對的序列先用 sliding window 擷取部分序列稱為 seeds;Align with FM index 就是字面上的意思,與先前建立好的 FM-index 進行初步比對,快速地僅針對 Seeds 與 FM-index 頭尾重複的部分,挑出所有可能的指定位置;Prioritize and resolve 透過逆向的 BWT 轉換揭露 reference 的原始面貌;Extend 階段利用 Single instruction multiple data (SIMD) 的 dynamic programming 來確認最終的指定位置。
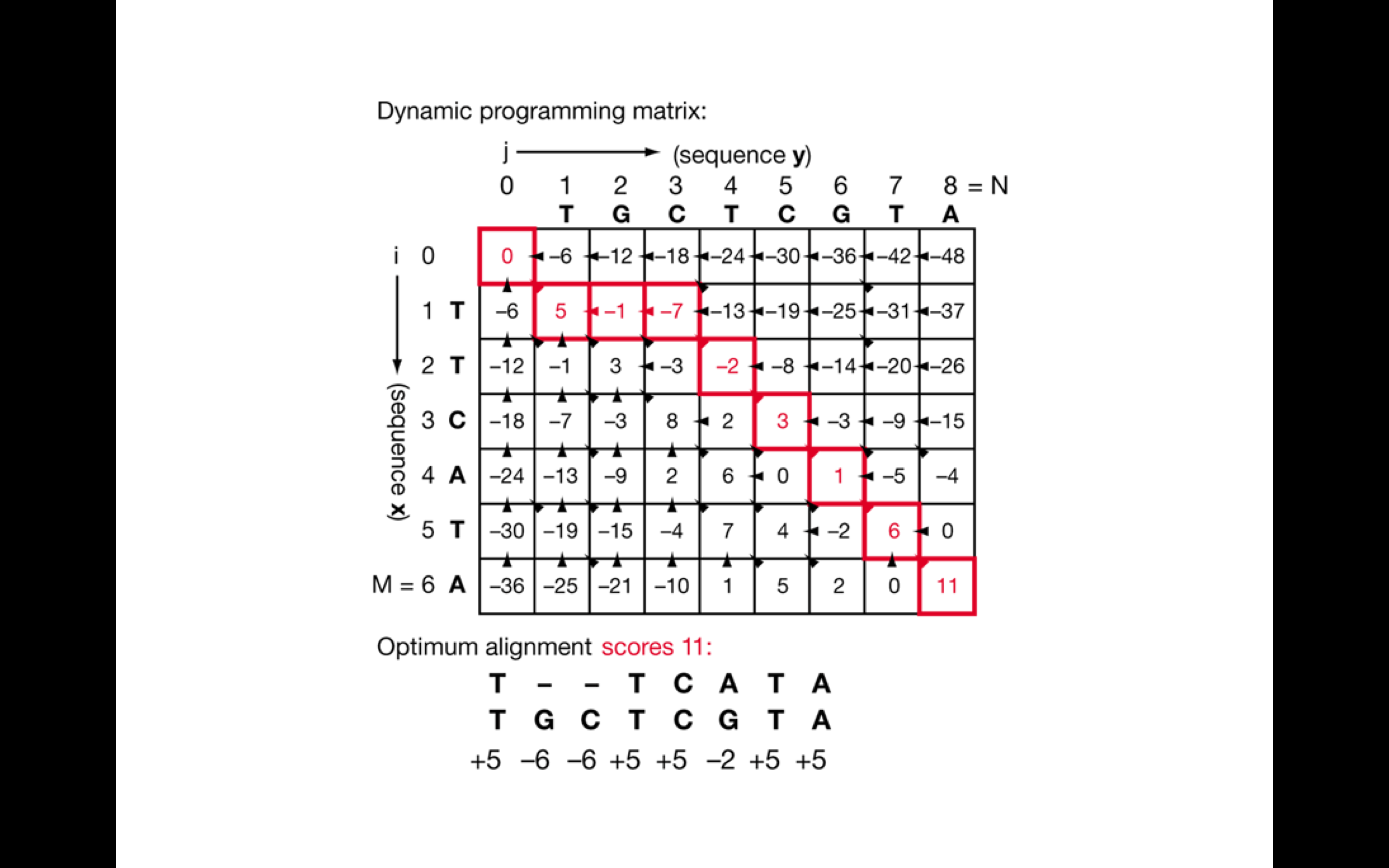
Dynamic programming 是一種以空間換取時間取得最佳解的方法。確認最終序列指定位置之後,軟體輸出 sequence alignment map (Sam) 檔案。
Bowtie2 實際使用及下游分析視覺化示範
下載已經編譯完成的檔案

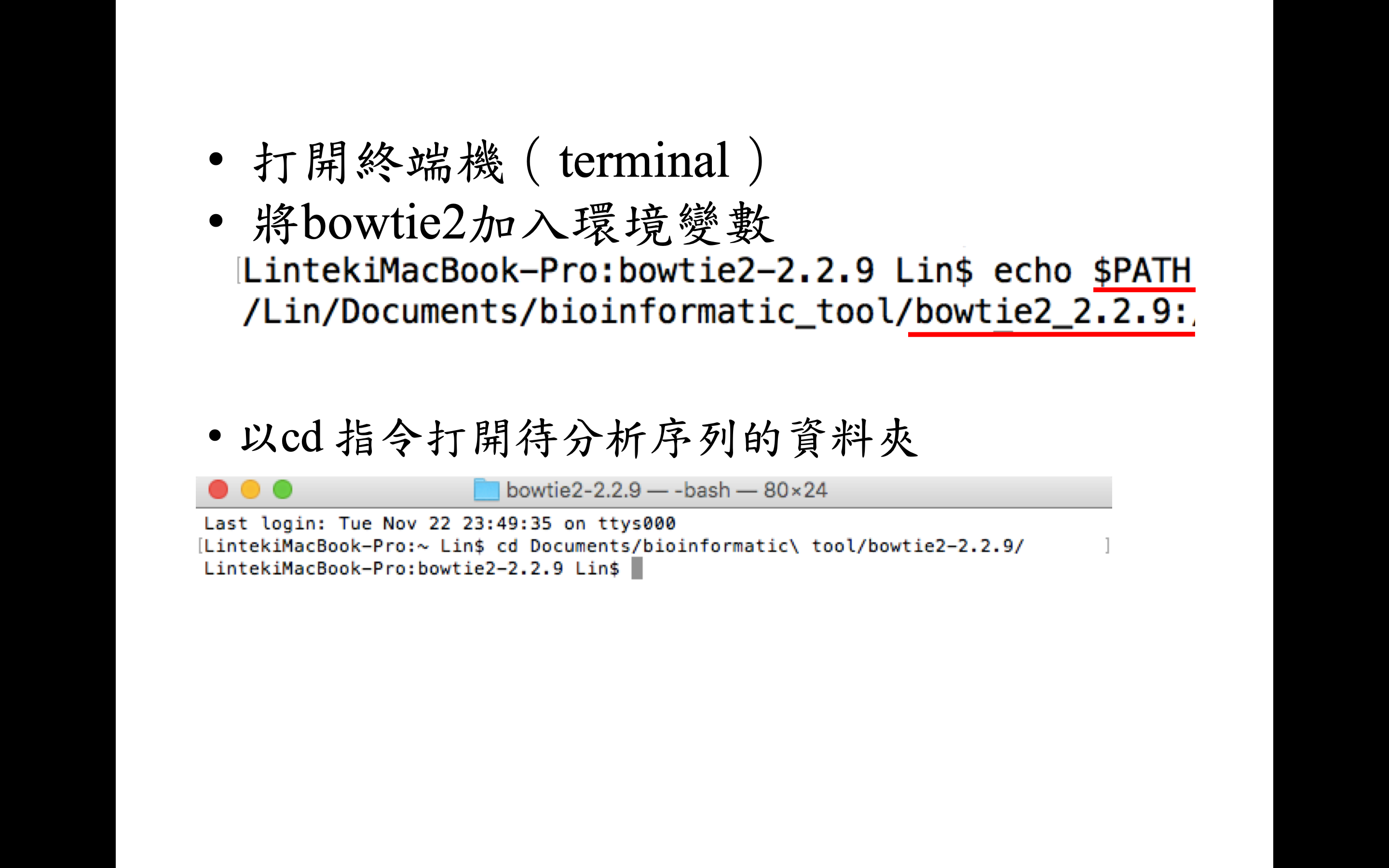
將 bowtie2 加入環境變數才可以隨處在終端機中呼叫指令
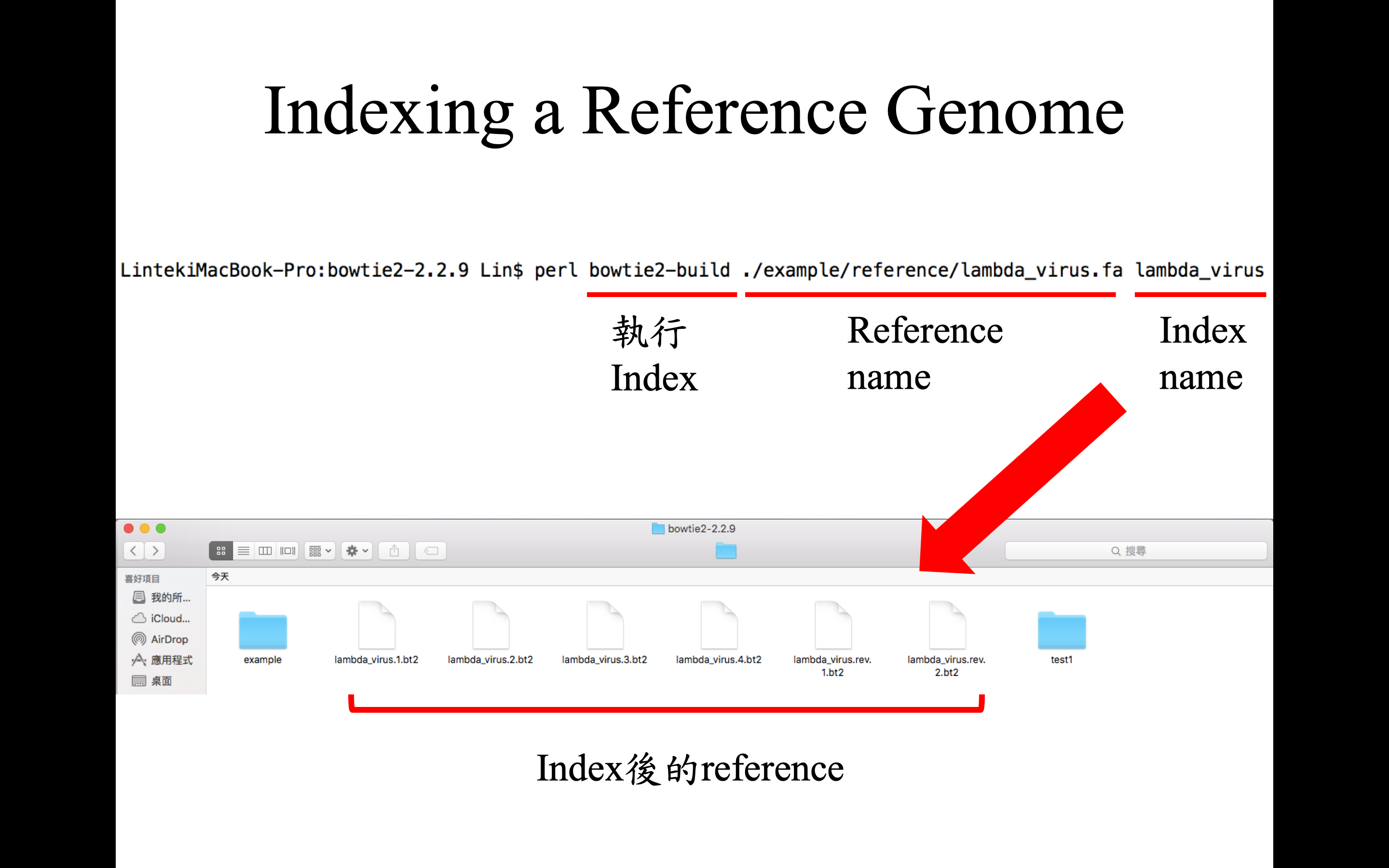
建立 index 指令,指令參數設置與 index 的樣子
實際 alignment 指令與終端機回傳資訊和輸出之檔案

以 Sublime text 3 開啟 SAM 檔案的樣子

回貼序列之後有幾種可能的下游分析

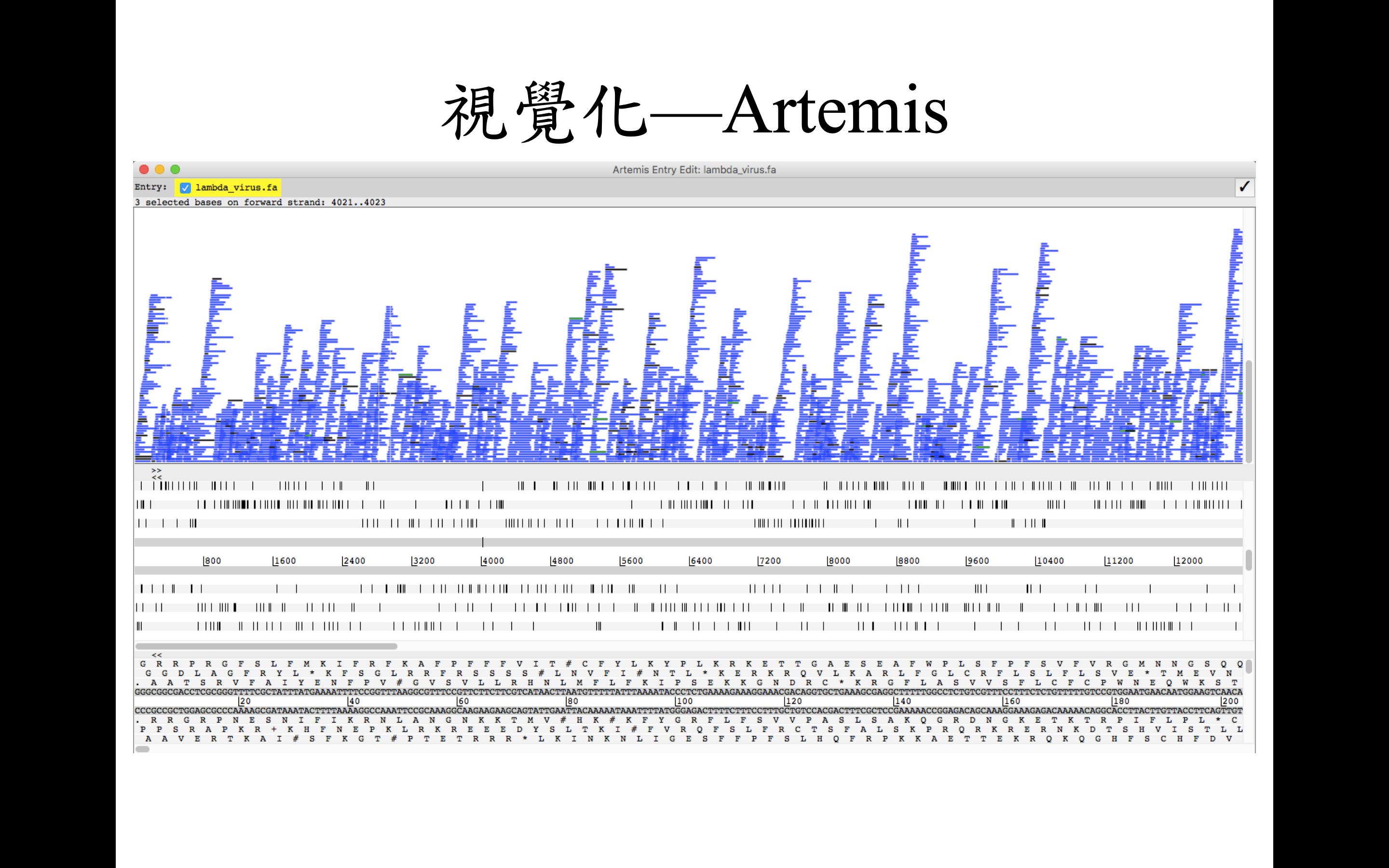
這邊先簡單地以兩款不同的軟體簡單示範回貼序列視覺化之後的樣子
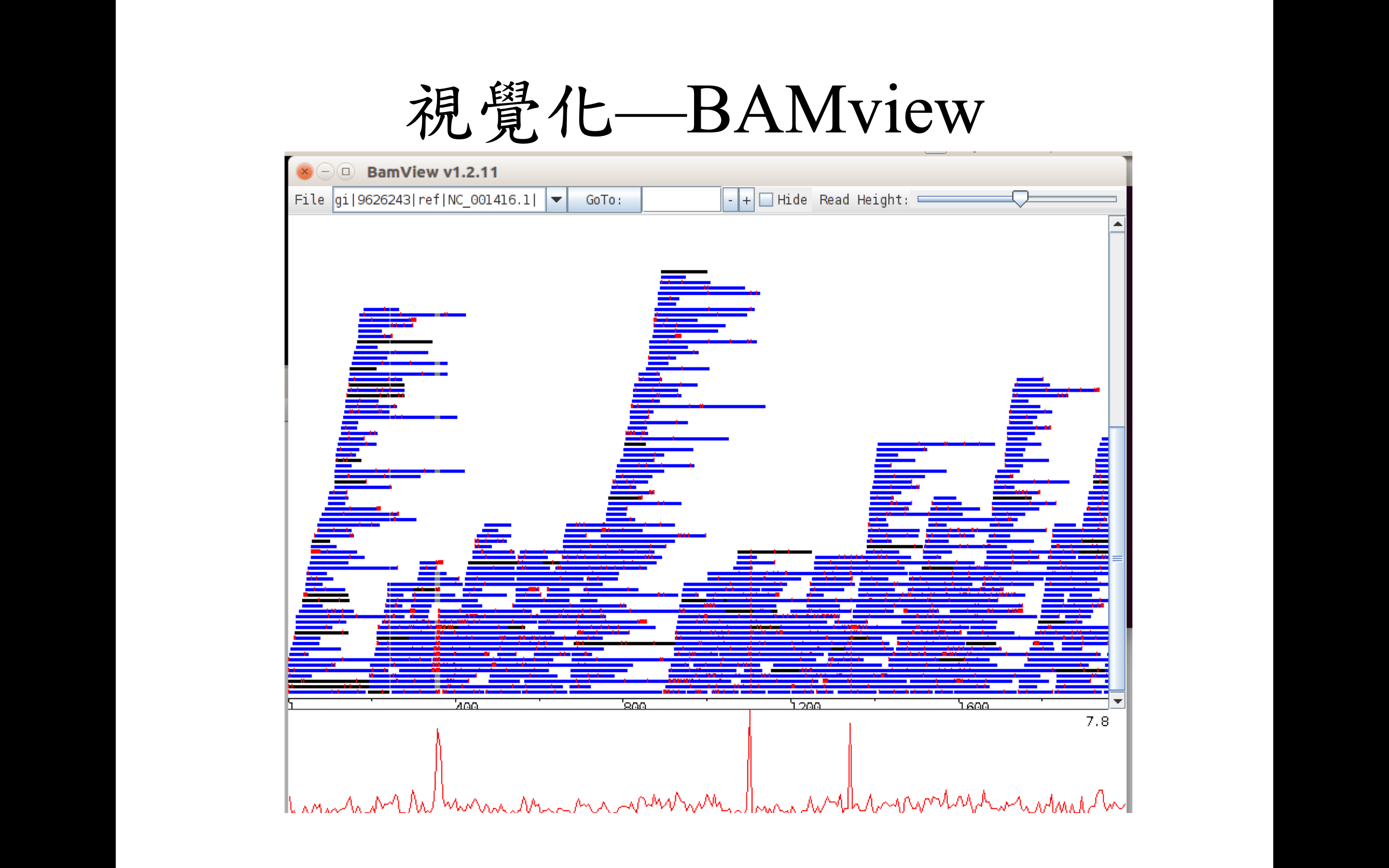
BAMview and Artemis
以上就是對 Bowtie2 的簡介,針對演算法與原理之處可能有錯誤之處,請不吝指教,如果有其他想要了解的軟體或細節請在下方留言~
參考文獻與延伸閱讀
Best RNA-Seq aligner: A comparison of mapping tools
Alignment-free sequence comparison: benefits, applications, and tools
Ultrafast and memory-efficient alignment of short DNA sequences to the human genome
註:本文部分圖片來自數年前謝晨與周林在基因體與系統生物學課堂報告之投影片。文中如果有未正確引用之處,請不吝告知,感謝!